Research Article
The Role of Tau Protein in Diseases

Bhaskar C Das1,2*, Sribidya Pradhan1, Devi Prasana Ojha1, Arpita Das1, Narayan S Hosmane3 and Sasmita Das4
1Departments of Medicine and Pharmacological Sciences, Icahn School of Medicine, Mount Sinai, New York 10029, USA2Department of Surgery, Weill Cornell Medical College of Cornell University, New York 10065, USA
3Department Chemistry and Biochemistry, Northern Illinois University, DeKalb, IL 60115, USA
4Nathan Kline Institute for Psychiatric Research, Orangeburg, NY 10962, USA
*Address for Correspondence: Bhaskar C Das, Departments of Medicine and Pharmacological Sciences, Department of Surgery, Weill Cornell Medical College of Cornell University, Icahn School of Medicine, Mount Sinai, New York 10029, USA, Email: [email protected]
Dates: Submitted: 22 March 2018; Approved: 06 April 2018; Published: 09 April 2018
How to cite this article: Das BC, Pradhan S, Ojha DP, Das A, Hosmane NS, et al. The Role of Tau Protein in Diseases. Ann Adv Chem. 2018; 2: 001-016. DOI: 10.29328/journal.aac.1001010
Copyright License: © 2018 Das BC, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Abstract
Amyloid-β peptide (Aβ) and tau protein deposits in the human brain are the pathological hallmarks of Alzheimer’s disease (AD). Tau is a class of proteins that are abundant in nerve cells and perform the function of stabilizing microtubules. However, in certain pathological situations, Tau proteins become defective and fail to adequately stabilize microtubules, which can result in the generation of abnormal masses that are toxic to neurons. This process occurs in a number of neurological disorders collectively known as Tauopathies. Tau protein is the major factor of the intracellular filamentous deposits that relate to a number of neurodegenerative diseases which includes the progressive supranuclear palsy (PSP), Pick’s disease, and Parkinsonism. The identification of mutations in Tau established that dysfunction or misregulation of tau protein is sufficient to cause dementia and neurodegeneration. In this review article, we discussed the etiology of the tau formation and role in AD and subsequently therapeutic approach for disassembling and Tau inhibition.
Introduction
The need for a new approach to treatment of Alzheimer’s disease is urgent. Alzheimer’s is the most common age-related dementia and the number of cases in the United States is expected to increase from the current number of about five to six million to 15 million by the year of 2050. The costs to family life and on the health care system are enormous. Alzheimer’s and other dementias are projected to cost the United States approximately $226 billion in 2015 alone (estimated 44.4 million, in 2013), with that number rising to as high as $1.1 trillion in 2050 (approx. 135.5 million in 2050) [1,2]. Tau protein is the major factor of the filamentous deposits that relate to AD alongside various other neurodegenerative disorders. Tau protein belongs to a group of proteins referred to as Microtubule-Associated Proteins (MAPs), that in common are heat resistant and limited affected by acid treatment without loss their function [3]. Six isoforms of tau protein differ according to the contents tubulin binding domains. These isoforms, which vary in size, are related to the presence or absence of sequences encoded by exons. Because each of these isoforms has explicit physiological roles, they are differentially expressed during the development of the brain. Tau has been said to interact with a number of other proteins besides tubulin/microtubules [4], although the biological relevance of many of these interactions is not clear.
Structure of Tau
Tau is an unusual protein that has long stretches of charged (positively and negatively) regions that are not beneficial for intermolecular hydrophobic association [5]. Of the four microtubule binding repeats in tau, the predicted amino acids having β-structure are concentrated. Tau is a hydrophilic protein that has been widely categorized in solution where, by analysis of the circular dichroism spectra [6], it appears as a random coiled protein. With its sedimentation coefficient, tau has been suggested to be a highly irregular protein, compatible with the long rod structure observed by electron microscopy [7]. Tau is encoded by a single gene, MAPT, which lies on human chromosome 17. In the human brain, tau is expressed as six molecular isoforms, which are the result of alternative splicing of exons 2, 3 and 10 in its pre-mRNA. The six isoforms of tau differ from one another in containing zero (0N), one (1N), or two (2N) amino-terminal inserts of 29 amino acids each, and the presence of three (3R) or four (4R) microtubule-binding domain repeats in a assumable length of 352 to 441 amino acids [8-11]. In the adults, it consists of a family of four to six related polypeptides with apparent mol. wts of 50,000 - 68,000 daltons [12].
Tau changes in the neuronal morphology
Neurons are cells with a very complex morphology; the microtubules become stabilized in specific directions that develop two types of cytoplasmic extensions that will become the axon and the dendrites, and tau is preferentially localized and active in distal portions of axons where they stabilize microtubules as well as providing flexibility. In Figure 1a,1b [9], the expression of tau in cells promotes the stabilization of microtubules, leading to the formation of cytoplasmic extensions as shown by the arrows. Neural transmission occurs through these processes, and any changes in neuronal morphology may affect the behavior and produce pathological events. In pathological situations, tau has additionally been shown to be capable of forming aberrant fibrillary polymers as shown in Figure 1c and Figure 2 (courtesy NRN poster 2008).
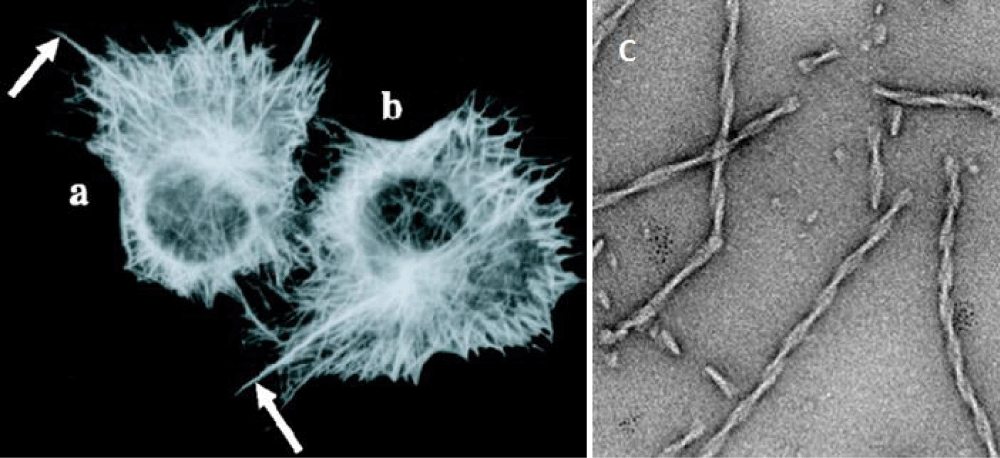
Figure 1: The expression of tau in non-neural cells (a and b) promotes the stabilization of microtubules, leading to the formation of cytoplasmic extensions (arrows) that are not normally seen in those cells that are not expressing tau. c: Fibers assembled in vitro from the pro-aggregant Tau repeat domain. (Courtesy [9] and modified by us).
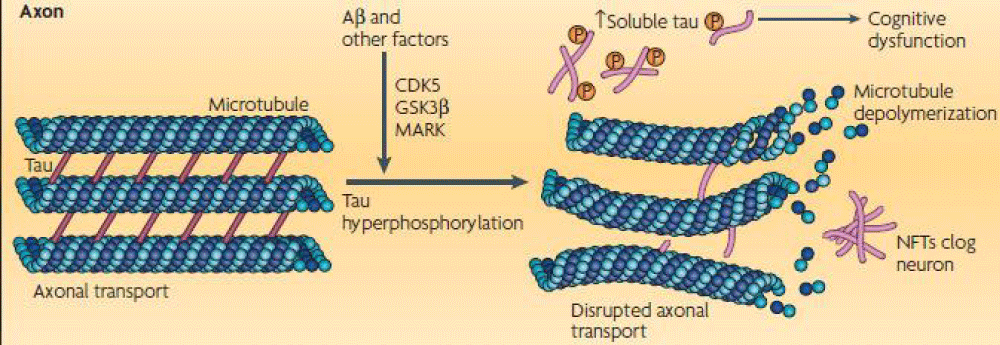
Figure 2: The image on the left shows a view of the binding of tau to a tubulin dimer. The right shows a description of the differences between a healthy neuron versus a diseased neuron and the result of a of the tau hyperphosphorylation. (Courtesy NRN poster 2008 and modified by us).
Formation of how Tau proteins
Tau proteins are produced through alternative splicing of a single gene called MAPT (Microtubule-Associated Protein Tau). The proteins work together with a spherical protein called tubulin to stabilize microtubules and aid the assembly of tubulin in the microtubules. Tau proteins achieve their control of microtubule stability through isoforms and phosphorylation. These six tau isoforms are generated by alternative mRNA splicing from a single gene in human brain [13,14]. Tau is a phosphoprotein that normally contains 2-3 phosphates/molecule, but it is abnormally hyperphosphorylated with a stoichiometry of 9-10 moles of phosphate per mole of protein in Alzheimer’s disease (AD) brain [15,16]. To date, more than 30 phosphorylation sites have been identified in AD hyperphosphorylated tau, some of which are not phosphorylated in normal tau. Hyperphosphorylation (mechanisms used by the cell to regulate mitosis) of tau proteins can cause the straight filaments to tangle which is referred to as neurofibrillary tangles. These tangles contribute to the pathology of Alzheimer’s disease. When a brain affected by Alzheimer’s disease is examined, all six isoforms of tau are often found hyperphosphorylated in paired helical filaments. Deposits of abnormal aggregates enriched with tau isoforms have also been reported in some other neurodegenerative diseases. Certain aspects of Alzheimer’s pathology also point at some similarities being shared with prion diseases.
Tau protein was discovered by studying factors necessary for microtubule formation. Tau protein promotes tubulin assembly into microtubules, one of the major components of the neuronal cytoskeleton that defines the normal morphology and provides structural support to the neurons [17,18]. Tubulin binding of tau is regulated by its phosphorylation state, which is regulated normally by coordinated action of kinases and phosphatases on tau molecule [19,20].
Taupathies
Tauopathies are considered as a group of disorders that are the consequence of abnormal tau phosphorylation, abnormal levels of tau, abnormal tau splicing, or mutations in the tau gene. In some tauopathies, like Alzheimer’s disease or Downs syndrome, the tau pathology is associated with other cerebral changes. The majority of neurodegenerative diseases are characterized by the deposition of insoluble protein in cells of the neuromuscular system. Advances in molecular neuropathology have allowed a classification system of neurodegenerative diseases based on this protein accumulation. Microtubule-associated tau is one protein that has important functions in healthy neurons, but forms insoluble deposits in diseases as tauopathies. Tauopathies encompass more than 20 clinicopathological entities, including Alzheimer’s disease, the most common tauopathy. There are important clinical, pathological, biochemical and genetic similarities in the range of these diseases and they have helped to advance our understanding of the factors that initiate neurodegeneration and tau accumulation.
Alzheimer disease is the most common and the best-studied tauopathy in which two main pathological structures form in the brains of patients: senile plaques (composed of the β-amyloid peptide), and neurofibrillary tangles (NFT), It generates problems in recent memory, a function associated to the temporal lobe, and in visual and spatial dysfunction and poor performance of over-learned tasks, functions of the parietal lobe. In connection with the global trend of prolonging human life and the increasing number of elderly in the population, the Alzheimer’s disease becomes one of the most serious health and socioeconomic problems of the present. Tau protein promotes assembly and stabilizes microtubules as mentioned before, which contributes to the proper function of neuron. Alterations in the amount or the structure of tau protein can affect its role as a stabilizer of microtubules as well as some of the processes in which it is implicated. Abnormal phosphorylation and truncation of tau protein have gained attention as key mechanisms that become tau protein in a pathological unit. Evidences about the clinic pathological significance of phosphorylated and truncated tau have been documented during the progression of AD as well as their capacity to use cytotoxicity when expressed in cell and animal models. Alzheimer’s disease is the most common type of dementia characterized by memory impairment and alteration of diverse cognitive abilities. Alzheimer’s disease and related tauopathies are histopathologically classified by slow and progressive neurodegeneration, which is associated mostly with intracellular buildup of tau protein leading to the neurofibrillary tangles (NFTs) and other inclusions containing modified tau (Figure 3) [21,22]. In pathological conditions, such as the case in AD, not only does abnormal phosphorylation of tau protein decrease its tubulin binding capacity leading to microtubule disorganization, but also this protein self-polymerizes and aggregates in the form of NFTs [21,22].
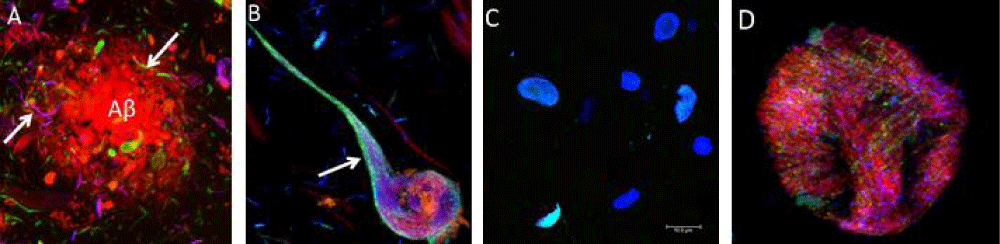
Figure 3: Courtesy [21] and modified by us) Histopathology of human brain [21,22]. A and B) Alzheimer´s disease, C) Pick’s dementia, C) corticobasal degeneration D) progressive supranuclear palsy.
In AD, the normal role of tau protein is ineffective to keep the cytoskeleton well organized in the axonal process because the protein loses its capacity to bind to microtubules. This abnormal behavior is endorsed by conformational changes and mis-folding in the normal structure of the tau [23-25] that leads to its aberrant aggregation into fibrillary structures inside the neurons of demented individuals [26-28]. Thus, most of the altered pools of tau protein in the disease are redistributed and aggregated in both the compartment and isolated processes of affected neurons. Alterations in the amount or the structure of tau protein can affect stabilization of microtubules and other processes related to this protein [29,30]. Furthermore, phosphorylated tau protein has attraction to the kinesin and therefore is transported to the distal sites of neuropil. This may account for the observation that tangle pathology in Alzheimer’s disease appears to initiate distally and then spreads in a reversing manner to the perikaryon. This process may be a mechanism to protect the stability of the microtubules by transporting hyperphosphorylated tau more rapidly to other cellular locations where tau can form aggregates [31].
Other diseases that are associated with tauopathy are Corticobasal Degeneration, Picks Disease, Progressive Supranuclear Palsy and Down syndrome. Corticobasal degeneration is a disorder characterized by cognitive disturbances like aphasia and apraxia, moderate dementia, and motor disturbances such as inflexibility, limb dystonia, and tremor. Pathological analyses have indicated glial and neuronal tau inclusions. Tau is also present in hyperphosphorylated form, but only certain molecular weight forms can be detected by electrophoresis. Pick’s disease is a dementia that produces disturbances in language and behavior and is associated with frontal lobe atrophy. It provokes changes in the character of the patient and in their interactions with others, as well as depression. This disorder is characterized by the presence of cytoplasmic tau inclusions in neurons of the frontal lobe, known as Pick bodies. The granular cells of the dentate gyrus are also affected. There is an absence of exon 10 expression [32], very vital component, suggesting that the degeneration of selective neuronal populations in different tauopathies reflects the physiological pattern of tau isoforms expressed. Progressive supranuclear palsy (PSP), also known as the Steele, Richardson, and Olszewski disorder, is characterized by supranuclear gaze palsy as well as by prominent postural instability, and in the later stages by dementia. Tau inclusions have been found in neuronal and glial cells, with both astrocytes and oligodendrocytes (coiled bodies) being affected [33]. Down syndrome is due to the trisomy of chromosome 21 that results in the defective growth and maturation of the brain, producing a cognitive impairment and dementia. In this disorder, tau is also hyperphosphorylated yielding a pattern similar to that of the Alzheimer’s disease. Other tauopathies involving hyperphosphorylated tau include Parkinsonism with dementia, myotonic dystrophy, and prion diseases with tangles.
Treatment options for Tauopathies
As of now, there are no approved and established pharmacologic treatment options for tauopathies. Available treatment strategies are based mainly on small clinical trials, miscellaneous case reports, or small studies. Approved therapeutic agents for Alzheimer´s dementia, such as acetylcholinesterase inhibitors and Memantine, have been used off-label to treat cognitive and behavioral symptoms in tauopathies, but the outcome has not been consistent. For behavioral or symptoms, treatment with antidepressants-especially selective serotonin reuptake inhibitors-could be helpful. In addition to pharmacologic treatment options, physical, or speech therapy can be applied to improve functional abilities. Each pharmacologic intervention should be fitted to the specific symptoms of the individual patient, and decisions about the type and duration of treatment should be based on its efficiency for the individual and the patient’s tolerance. Currently, no effective treatment is available that targets the cause of these diseases. Overall, only symptomatic treatment is available for AD and tau protein misfolding diseases, and new therapeutic concepts range from tau vaccinations and antiphosphorylation strategies to microtubule-stabilizing and antiaggregation drugs.
Anti-inflammatory or antioxidant drugs based impairments
Based on clinical observations, the individuals with treatment of Epidemiological studies were chronically maintained on anti-inflammatory or antioxidant drugs relevant other disease conditions had lower risks for developing cognitive impairment and AD [34]. The hypothesis was approved by various other studies which shows the decrease of NFTs [35,36], tau phosphorylation [37], neurobehavioral (memory) deficits [38], Aβ42 burden [39,40], synaptic degeneration [41-43], and mitochondrial dysfunction [44]. Various anti-oxidants and anti-inflammatory agents that have provided neuroprotection for AD are curcumin [40], alpha-lipoic acid [45-47], Vitamin E [48,49], resveratrol [50], phytonutrients [51], cyclooxygenase 2 inhibitors [52], Ginkgo [53], and melatonin [54].
Insulin therapy
Studies have found that the major role for a tau hyperphosphorylation which leads to deformation of tau proteins, can be attributed to (i) central disruption of insulin signaling by insulin receptor or (ii) insulin receptor substrate (IRS) 2 gene deletions, (iii) following intracerebral streptozotocine injections (Figure 4) [55]. Therefore, an insulin supplement could improve the cognition and brain energy metabolism in people with mild cognitive impairment or early AD. Further, studies have found that insulin administration enhances clearance of Aβ-42, decreases activity of kinases that promote tau hyperphosphorylation, and enhances signaling through pathways needed for synaptic plasticity [55-58].
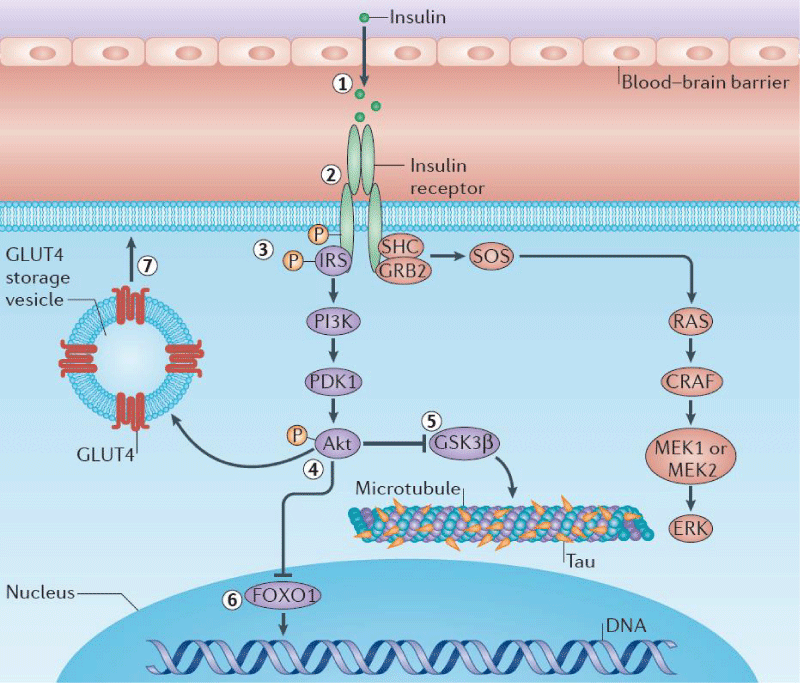
Figure 4: Relevance of tau and Insulin pathway. [55-58] (Courtesy [55] and modified by us).
Therapeutics
a) Tau anti-phosphorylation
In another mechanism, the inhibition of hyperphosphorylation could produce an avoidable condition for the formation of the neurofibrillary tangles. Studies have shown that the glycogen synthase kinase-3 (GSK-3) protein has a prominent function in the stimulation of the hyperphosphorylation of tau proteins. W. Noble and K. Duff presented [59] an in vivo study with transgenic mice overexpressing mutant human tau. The mice were treated with the glycogen synthase kinase-3 (GSK-3) inhibitor lithium chloride. Treatment resulted in significant inhibition of GSK-3 activity. Lithium administration also resulted in significantly lower levels of phosphorylation at several epitopes of tau known to be hyper phosphorylated in Alzheimer’s disease and significantly reduced levels of aggregated, insoluble tau. The study also found that the lithium-chloride-treated mice showed less degeneration if administration was started during early stages of tangle development. These results support the idea that kinases are involved in tauopathy progression and that kinase inhibitors may be effective therapeutically [59]. Potential therapies primarily involve deformed tau proteins. These therapeutic targets include inhibition of tau hyperphosphorylation, microtubule stabilization, prevention of tau oligomerization, and enhancement of tau degradation as well as tau immunotherapy. With no cure till date, however the important part is the few drugs are in preclinical trials. One of such promising drug is LMTX (Scheme 1), which is currently in the third phase of clinical trials [60]. The methylthionium chloride derived LMTX, generally acts as an inhibitor of tau hyperphosphorylation.
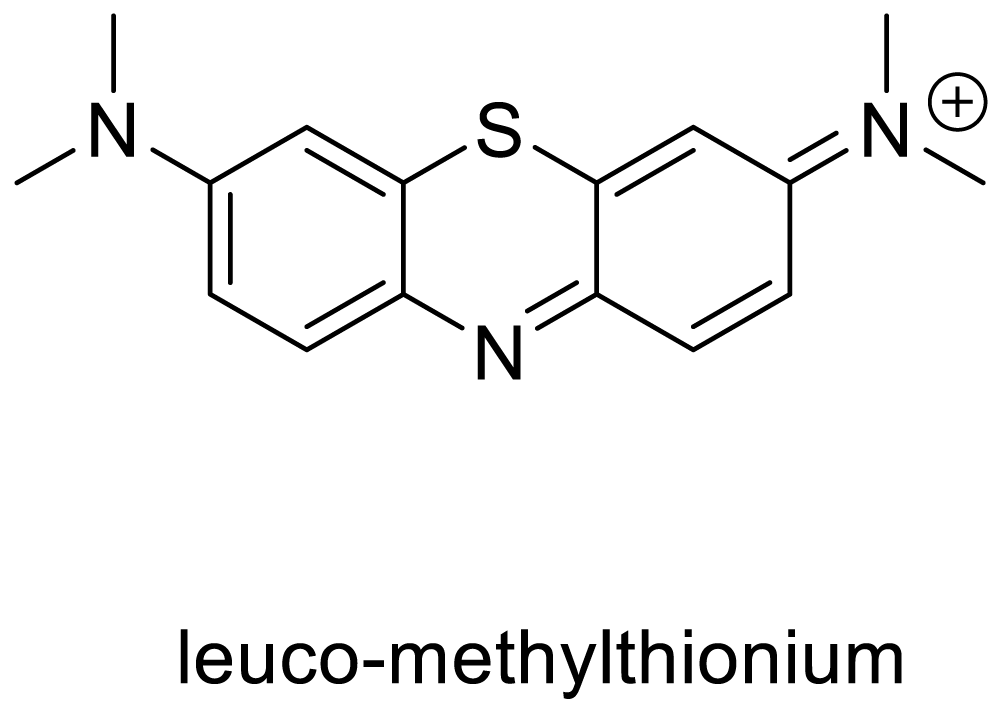
Scheme 1: Chemical structure of LTMX drug.
b) Tau Dephosphorylation
The counteraction mechanism against the tau anti phosphorylations would be dephosphorylation process, which can be regenerating the tau proteins from the toxic, and aggregated Paired helical filaments (PHFs). In a process to cure the hyperphosphorylated proteins, Gong et al studied the Kinetics of Tau Dephosphorylation Catalyzed by PP5. PP5 is a phosphoseryl/phosphothreonyl protein phosphatase with an effective character of tau dephosphorylation studied previously by the same groups [63]. Interestingly, PP5 dephosphorylates tau at least at 12 phosphorylation sites with different efficiencies. Further, PP5 can dephosphorylate tau in living cells with AD brain. Overall the studies are in progress to provide dephosphorylation could be a possible strategy in curing AD affected brains [64]. On basis of few reports, both agonists and antagonists of 5HT6 have crucial impact over improving memory and learning in humans as well as animals. It is also evident that the cholinergic activity gradually deteriorates, due to degeneration of cholinergic neurons. Therefore, compounds affecting 5-HT6 receptors represent potential therapeutic targets for symptomatic treatment of AD. In this concern, the Idalopirdine (Lu AE58054) acts as a selective 5HT6 receptor antagonist and has shown tremendous clinical phase II trials with little side effects. Further the drug is in phase III trials [65-68]. In a similar mechanism of function the drug SB742457 (Scheme 3) could produce effective results compared to the idalopirdine [69].
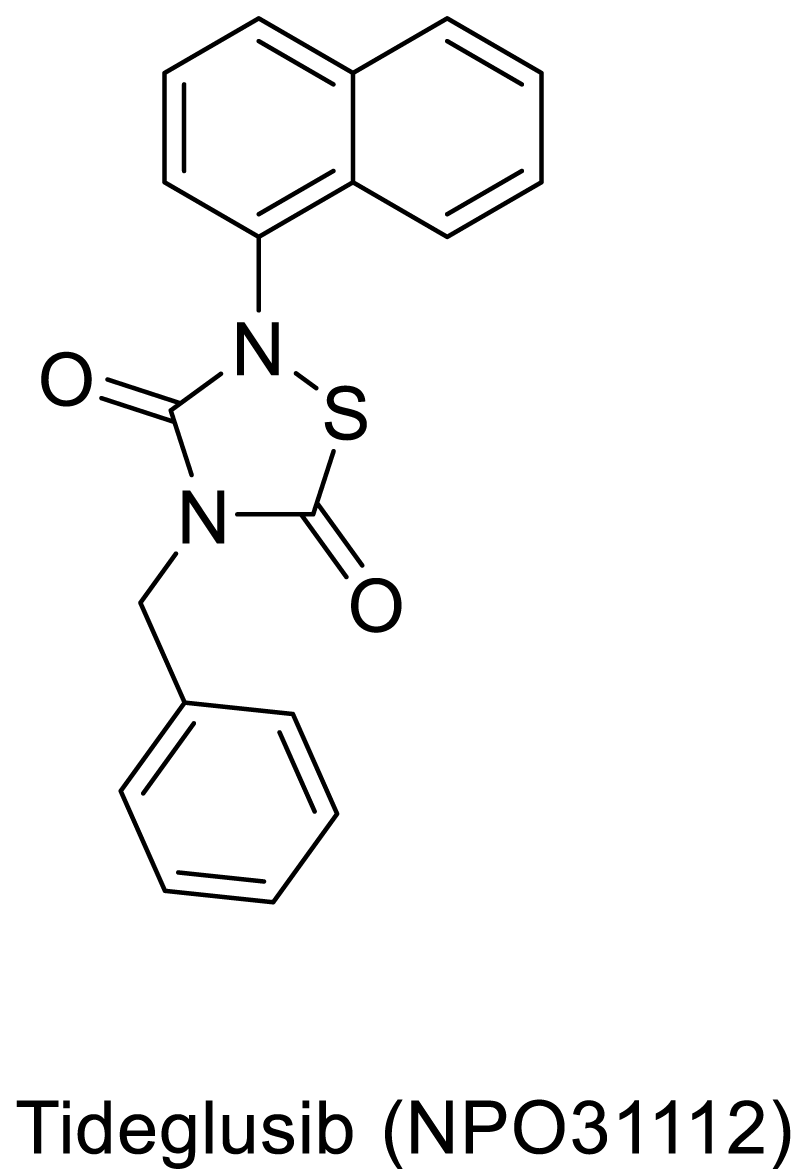
Scheme 2: Tau anti-phosphorylation drug Tideglusib.
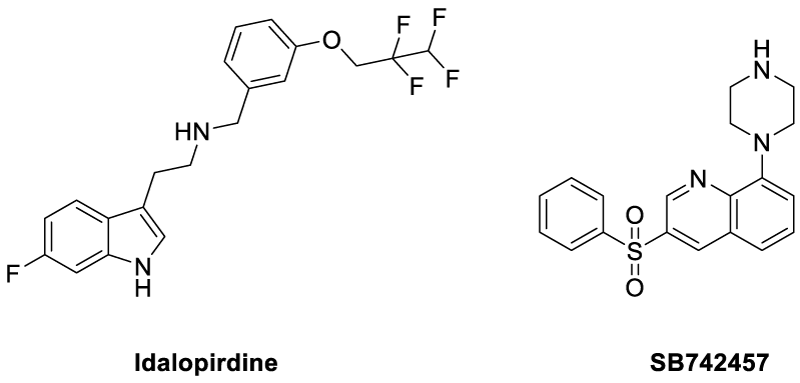
Scheme 3: Chemical structure of 5HT6 receptor antagonists.
Similar to the serotonin receptors, the presynaptic histamine H3 receptors also are evenly found in the prefrontal cortex, hippocampus, and hypothalamus. Therefore are equally responsible for the memory and recognition aspects for the human brain. The use of H3 antagonists are expected to enhance the cholinergic neurotransmission as a result of release of acetylcholine, dopamine and GABA into the synaptic cleft. Hence working on this hypothesis two drugs were introduced, such as ABT-288 and GSK239512 (Scheme 4). The phase I and phase II clinical trials of the former one have shown tremendous anti-AD activity while the later drug has not shown any considerable progress in low-moderate AD patients [70-73].
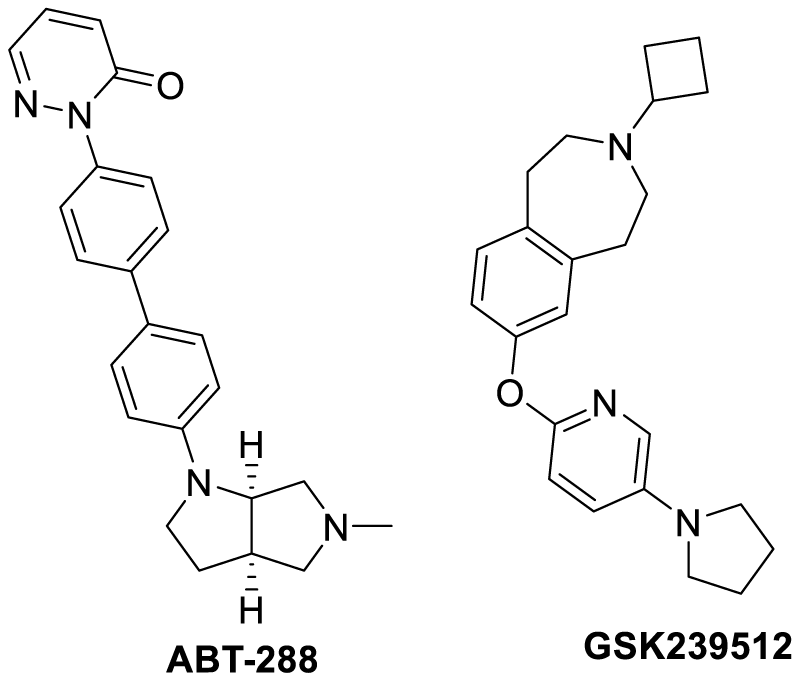
Scheme 4: Chemical structure of histamine H3 receptor antagonists.
Azeliragon (TTP488) is an oral small-molecule inhibitor of the receptor for advanced glycation endproduct (RAGE), which mediates transport of the Aβ peptide from peripheral to CNS compartments in the brain [74]. This drug is currently undergoing phase III trials in patients with mild-to-moderate AD (NCT02080364). Encenicline has been introduced into phase III trials with a positive results in phase II. The drug is a selective α7-nAChR (a-7 nicotinic acetylcholine receptor) agonist. Similarly Nilvadipine is a dihydropyridine calcium antagonist currently approved for treatment of hypertension (Nivadil) (Scheme 5). Clinical observations have revealed its positive influence on cognition in treated patients [75,76].
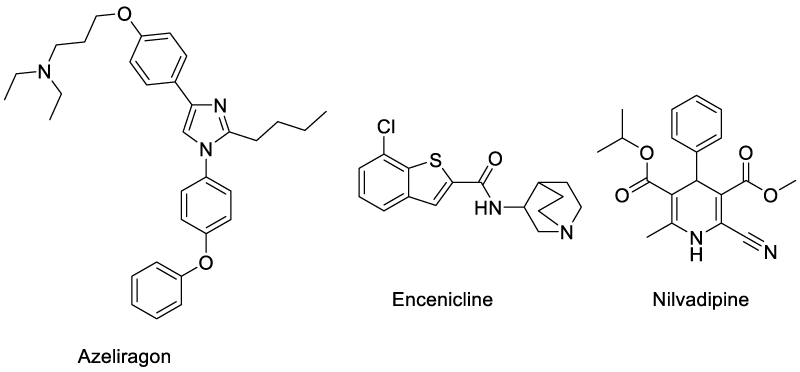
Scheme 5: Structure of anti-Alzheimer’s drugs in phase III clinical trials representing different mechanisms of action.
c) Metal Chelators: A Possible Chemotherapy for Alzheimer’s Disease
Distribution of copper, iron and zinc as the key components connected to amyloid metabolism. Therefore the dysfunctions of these metal ions are responsible for the deposits of amyloids and other misfolded proteins resulting in the various neurodegenerative diseases. The restoration of these metal ions homeostasis in any form can possibly be an essential chemotherapeutic. External addition of few chelators has found improved activity for the metal ions. These stimulates by a weak coordination to the metal ion and further bind to the protein or oligomer deposits. In such a case, the potential of PcTS (Scheme 6) as an amyloid modulator was explored on the protein tau and the beta-amyloid (Aβ) peptide, considered as the pathological components of Alzheimer’s disease. PcTS was able to interfere with tau filament assembly in vitro by inducing the conversion of the nontoxic protein state into soluble oligomers characterized by the lack of α-helix or β-sheet structural elements in its molecular architecture [77].
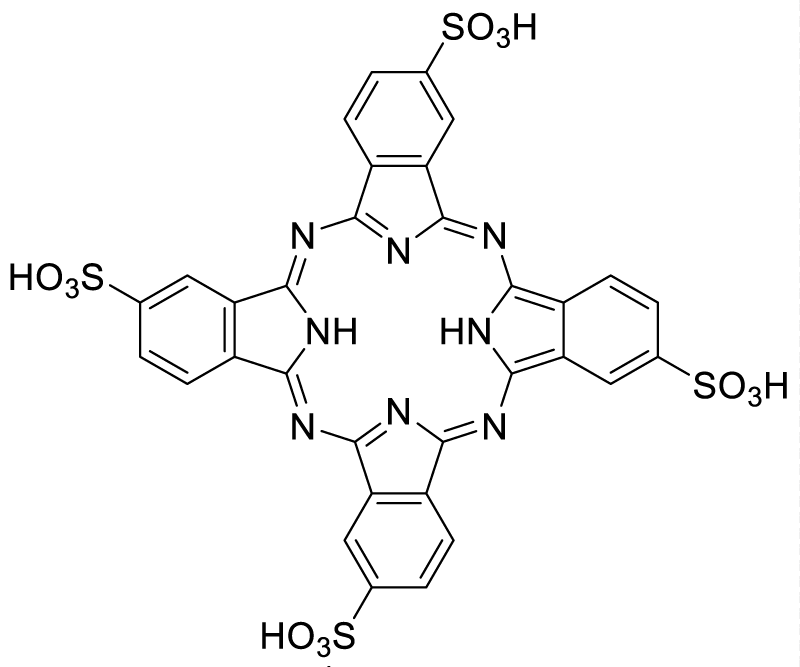
Scheme 6: Chemical structure of PcTS as an amyloid modulator.
d) Tau Vaccination
The amyloid cascade hypothesis postulates that Aβ deposition is an initial event in the pathogenesis and Aβ deposition may precede AD symptoms in some patients by at least 20 years. Amyloid-β therapy with active and passive immunizations against Aβ has a high possibility to be effective in removing Aβ from brain and might prevent the downstream pathology. Since 2000, a number of clinical trials for AD immunotherapy have started, have failed, and are continuing to be pursued. Asuni and coworkers studied a vaccination of P301L transgenic mice, with developed neurofibrillary tangles in several brain regions and the spinal cord, with a tau fragment peptide. Vaccination reduced aggregated tau in the brain and slowed progression of the tangle-related behavioral phenotype compared with control animals [78].
e) Microtubule Stabilizing by Therapeutics
Normal tau has a key role in stabilizing the microtubules and maintaining MT networks essential for axonal transport in neurons. The failure of these actions by the hyper phosphorylated tau results in the instability of microtubules and are characterized as tau inclusions. Trojanowski JQ et al., have studied a microtubule-binding by external drugs which could facilitate the tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. The finding employed paclitaxel or Taxol (well-known anti-cancer drug) injected into transgenic mice for a period of 12 weeks. The study showed that the results were interesting as both the stable microtubules were increased and also restored fast axonal transport in spinal axons [79]. In another case, the stabilization of microtubules by tubulin binding had been approached with a small molecule drug epothilone D (BMS-241027) and TPI-287 (Scheme 7) [80].
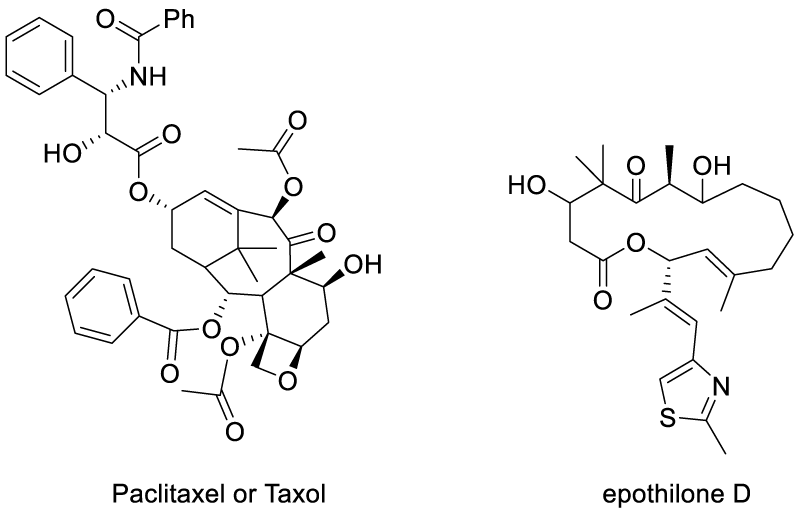
Scheme 7: Chemical structure of Paclitaxel or Taxol.
f) Cell Therapy
In the last few years, hopes were raised that cell replacement therapy would provide cure by compensating the lost neuronal systems. Stem cells obtained from embryonic and adult tissue and grafted into the intact brain of mice or rats were mostly followed by their incorporation into the host parenchyma and differentiation into functional neural lineages. In the lesioned brain, stem cells exhibited targeted migration towards the damaged regions of the brain, where they engrafted, multiplied and matured into functional neurons. Observations in animal models of AD have provided evidence that transplanted stem cells or neural precursor cells (NPCs) survive, migrate, and differentiate into cholinergic neurons, astrocytes, and oligodendrocytes with amelioration of the learning/memory deficits. Besides replacement of lost or damaged cells, stem cells stimulate endogenous neural precursors, enhance structural neuroplasticity, and down regulate pro-inflammatory cytokines and neuronal apoptotic death. Stem cells could also be genetically modified to express growth factors into the brain. In the last years, evidence indicated that the adult brain of mammals preserves the capacity to generate new neurons from neural stem cells. Interestingly, many drugs including antidepressants, lithium, acetyl cholinesterase and inhibitors were able to enhance the impaired neurogenesis in this disease process. This paved the way towards exploring the possible pharmacological manipulation of neurogenesis which would offer an alternative approach for the treatment of AD [81-87].
g) Essential Fatty Acids and Tau in Neurodegenerative Diseases
Polyunsaturated fatty acids (PUFA) are required for maintaining the structure, function and vascular integrity of the brain. The brain can synthesize non-essential fatty acids, but essential PUFA (e.g., arachidonic acid [AA] and docosahexaenoic acid [DHA]) are largely acquired from the peripheral circulation [88]. Arachidonic acid metabolism produces pro-inflammatory lipid metabolites, such as prostaglandins and leukotrienes, whereas metabolism of DHA generates anti-inflammatory mediators, such as resolvins. Hence, an increase in AA to DHA ratios could promote inflammation, further contributing to AD pathogenesis [89]. Docosahexaenoic acid (DHA, 3 22:6, n-3) is an n-3 long-chain PUFA that has recently attracted attention because of its various beneficial effects on human health. DHA is essential for brain development, and DHA or its precursors, such as α-linolenic acid (18:3, n-3) must be obtained from the diet or from the maternal supply during pregnancy because α-linolenic acid cannot be synthesized de novo by animal tissues. DHA accumulation in neural tissue is increased greatly during the perinatal period, when the frontal lobe and hippocampus undergo intensive growth and neurogenesis, neuritogenesis, and synaptogenesis occur actively. Therefore, an adequate supply of DHA is required for proper brain development [90].
h) The role of Retinoic Acid (RA) in Tau modulation
Retinoic acid, an essential factor derived from vitamin A, has been shown to have a variety of functions including roles as an antioxidant and in cellular differentiation. Since oxidative stress and de-differentiation of neurons appear to be common pathological elements of a number of neurodegenerative disorders, RA may offer therapeutic promise. RA may have therapeutic properties ideally served for the treatment of AD. Dosage and safety monitoring will be a major challenge to focus on primarily. An expectation is there of line of clinical trials to arise in the near future, which will be designed to better understand the effectiveness of the mechanisms with RA. Very recently Das and his group published review articles elucidating the role of natural and synthetic retinoids in AD [91,92].
Etiology
The etiological factors, other than older age and genetic susceptibility, remain to be determined. Increasing evidence strongly points to the potential risk roles of vascular risk factors and disorders for example smoking, midlife high blood pressure and obesity, diabetes, and cerebrovascular lesions and the possible beneficial roles of psychosocial factors such as higher education, social engagement, physical exercise, and mentally stimulating activities, in the pathogenetic process and clinical manifestation of the dementing disorders. The interventions of vascular risk factors and the maintenance of socially integrated lifestyles and mentally stimulating activities are expected to reduce the risk of the clinical onset of dementia, including AD.
Summary
Tau is a microtubule-associated protein that stabilizes neuronal microtubules under normal physiological conditions due to functional redundancy and the presence of other microtubule-associated proteins. However, in pathological situations tau may be hyperphosphorylated and assembled in an aberrant way. As a consequence of these modifications, neural toxicity supplements, resulting in the appearance of neurological disorders, mainly dementias for example the Alzheimer’s disease, which are collectively known as tauopathies. Different types of neurons are damaged in different tauopathies, and the outline of the mechanisms underlying the basis for this specificity will probably be the objective neurodegeneration.
It is largely accepted that clinical appearance of dementia in Alzheimer’s disease is due to the neuronal loss occurring in those areas of the brain associated with cognitive functions of the patients. Fibrillary presences are reported to be responsible for cell death. However, inconsistency has emerged from studies demonstrating that cognitive impairment in animal models occurs earlier than the initial formation of fibrillary structures. In this concern, numerous of reports analyzing the brain of Alzheimer diseased patients come to an agreement that fibrillary aggregation of tau is the best correlator with the onset and progression of dementia. It is mostly accepted that abnormal posttranslational modifications, that is, hyperphosphorylation, acetylation, glycation, nitration, truncation, and others, are responsible for altered tau structure in Alzheimer’s disease. Moreover, accurate determination of altered tau protein in the cerebrospinal fluid and other body fluids may provide better expectation to predict the onset and evolution of dementia.
References
- Wimo A, Jonsson L, Bond J, Prince M, Winblad B, et al. Alzheimer Disease international. The worldwide economic impact of dementia 2010. Alzheimers Dement. 2013; 9: 1-11. https://goo.gl/A1pLaU
- Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol.2011; 7: 137-152. Ref.: https://goo.gl/eNPeUJ
- (a) Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Bio. 1977; 116: 227-247. Ref.: https://goo.gl/KEAE1k; (b) Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017; 133: 665-704. Ref.: https://goo.gl/26pR2Z; (c) Avila J, Lucas JJ, Pérez M, Hernández F. Role of Tau Protein in both Physiological and Pathological Conditions. Physiol Rev. 2004; 84: 361-384. Ref.: https://goo.gl/v86s1E; (d) Nadimidla K., Ismail T., Kanapathipillai M., Tau peptides and tau mutant protein aggregation inhibition by cationic polyethyleneimine and polyarginine. Biopolymers. 2017; 107: 23024. Ref.: https://goo.gl/Vmmzn1
- Morris M, Maeda S, Vossel K, Mucke L. The Many Faces of Tau. Neuron. 2011; 70: 410-426. Ref.: https://goo.gl/1ukURr
- Ruben GC, Iqbal K, Grundke-Iqbal I, Wisniewski HM, Ciardelli TL, et al. The microtubule associated protein tau forms a triple-stranded left-hand helical polymer. J Biol Chem. 1991; 266: 22019-22027. Ref.: https://goo.gl/pzTXA6
- Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977; 116: 207-225. Ref.: https://goo.gl/HyVHN9
- Imahori K, Uchida T. Physiology and pathology of tau protein kinases in relation to Alzheimer's disease. J Biochem. 1997; 121: 179-188. Ref.: https://goo.gl/JxvEpC
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989; 8: 393-399. Ref.: https://goo.gl/j1xdLw
- Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol. 1986; 103: 2739-2746. Ref.: https://goo.gl/Mt6JGU
- Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, et al. Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci. 2004; 24: 2304-2312. Ref.: https://goo.gl/rxKLHg
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, et al. Amyloid‑β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011; 31: 700-711. Ref.: https://goo.gl/fLfrKo
- Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977; 116: 207-225. Ref.: https://goo.gl/KaxvbQ
- Goedert M, Spillanti MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989; 3: 519-526. Ref.: https://goo.gl/KsoMmD
- Himmler A. Structure of the bovine tau gene: alternatively spliced transcripts generate a protein family. Mol Cell Biol. 1989; 9: 1389-1396. Ref.: https://goo.gl/ZCX9yT
- Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, et al. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993; 268: 24374-24384. Ref.: https://goo.gl/mq6KFV
- Kenessey A, Yen SH. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 1993; 629: 40-46. Ref.: https://goo.gl/xRhuAJ
- Maccioni RB, Muñoz JP, Barbeito L. The molecular bases of Alzheimer's disease and other neurodegenerative disorders. Arch Med Res. 2001; 32: 367-381. Ref.: https://goo.gl/ukHCSD
- Kosik KS. The molecular and cellular biology of tau. Brain Pathol. 1993; 3: 39-43. Ref.: https://goo.gl/SZBXE6
- Mandelkow EM, Biernat J, Drewes G, Gustke N, Trinczek B, et al. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging. 1995; 16: 355-363. Ref.: https://goo.gl/4rCGDU
- Liu F, Iqbal K, Grundke-Iqbal I, Rossie S, Gong CX. Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer's disease. J Biol Chem. 2005; 280: 1790-1796. Ref.: https://goo.gl/KNZjXj
- Avila J. Tau kinases and phosphatases: commentary. J Cell Mol Med. 2008; 12: 258-259. Ref.: https://goo.gl/Xt3StM
- Iqbal, Grundke-Iqbal I. Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention: Alzheimer review series. J Cell Mol Med. 2008; 12: 38-55. Ref.: https://goo.gl/C748JD
- Carrell RW, Gooptu B. Conformational changes and disease-serpins, prions and Alzheimer's. Curr Opin Struct Biol. 1998; 8: 799-809. Ref.: https://goo.gl/Ss93aG
- Fox N, Harvey RJ, Rossor MN. Protein folding, nucleation phenomena and delayed neurodegeneration in Alzheimer's disease. Rev Neurosci. 1996; 7: 21-28. Ref.: https://goo.gl/85dSQN
- Hyman BT, Augustinack JC, Ingelsson M. Transcriptional and conformational changes of the tau molecule in Alzheimer's disease. Biochim Biophys Acta. 2005; 1739: 150-157. Ref.: https://goo.gl/h7mMjT
- García-Sierra F, Ghoshal N, Quinn B, Berry RW, Bínder LI. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimers Dis. 2003; 5: 65-77. Ref.: https://goo.gl/JxY4ZH
- Ghoshal N, García-Sierra F, Fu Y, Beckett LA, Mufson EJ, et al. Tau-66: evidence for a novel tau conformation in Alzheimer’s disease. J Neurochem. 2001; 77: 1372-1385. Ref.: https://goo.gl/GkAmVcg
- Ghoshal N, García-Sierra F, Wuu J, Leurgans S, Bennett DA, et al. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer's disease. Exp Neurol. 2002; 177: 475-493. Ref.: https://goo.gl/NC62Zm
- Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003; 24: 1079-1085. Ref.: https://goo.gl/BAbCfL
- LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, et al. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009; 87: 440-451. Ref.: https://goo.gl/JwWhxM
- Cuchillo-Ibanez I, Seereeram A, Byers HL, Leung KY, Ward MA, et al. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008; 22: 3186-3195. Ref.: https://goo.gl/9PqPpd
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989; 8: 393-399. Ref.: https://goo.gl/14gouX
- Delisle MB, Murrell JR, Richardson R, Trofatter JA, Rascol O, et al. A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol. 1999; 98: 62-77. Ref.: https://goo.gl/6PDHpy;
- (a) Standridge JB. Pharmacotherapeutic approaches to the prevention of Alzheimer’s disease. Am J Geriatr Pharmacother. 2004; 2: 119-132. Ref.: https://goo.gl/FtcQSa; (b) XieYibin Y, Youbiao T, Xiubo Z, Du X, Liu Q. Ebselen ameliorates β-amyloid pathology, tau pathology, and cognitive impairment in triple-transgenic Alzheimer’s disease mice. J Biol Inorg Chem. 2017; 22: 851-865. Ref.: https://goo.gl/V5HQ8t ; (c) Dioli C, Patrício P, Trindade R, Pinto LG, Silva JM, et al. Tau-dependent suppression of adult neurogenesis in the stressed hippocampus. Mol Psychiatry. 2017; 22: 1110-1118. Ref.: https://goo.gl/GVLBH2
- Boimel M, Grigoriadis N, Lourbopoulos A, Touloumi O, Rosenmann D, et al. Statins reduce the neurofibrillary tangle burden in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009; 68: 314-325. Ref.: https://goo.gl/mFyk4e
- Piedrahita D, Hernandez I, Lopez-Tobon A, Fedorov D, Obara B, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s mice. J Neurosci. 2010; 30: 13966-13976. Ref.: https://goo.gl/jY9YpV
- Leroy K, Ando K, Heraud C, Yilmaz Z, Authelet M, et al. Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J Alzheimers Dis. 2010; 19: 705-719. Ref.: https://goo.gl/j8Cc2M
- Villaflores OB, Chen YJ, Chen CP, Yeh JM, Wu TY. Curcuminoids and resveratrol as anti-Alzheimer agents. Taiwan J Obstet Gynecol. 2012; 51: 515-525. Ref.: https://goo.gl/dUtiou
- Lazar AN, Mourtas S, Youssef I, Parizot C, Dauphin A, et al. Curcumin-conjugated nanoliposomes with high affinity for Abeta deposits: possible applications to Alzheimer disease. Nanomedicine. 2013; 9: 712-721. Ref.: https://goo.gl/TKJuPP
- Hamaguchi T, Ono K, Yamada M. REVIEW: Curcumin and Alzheimer’s disease. CNS Neurosci Ther. 2010; 16: 285-297. Ref.: https://goo.gl/jz9Ykm
- Malm T, Mariani M, Donovan LJ, Neilson L, Landreth GE. Activation of the nuclear receptor PPARdelta is neuroprotective in a transgenic mouse model of Alzheimer’s disease through inhibition of inflammation. J Neuroinflamm. 2015; 12: 7. Ref.: https://goo.gl/Xuqsc7
- Olmos-Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain. 2016; 139: 891-907. Ref.: https://goo.gl/Nb7PLm
- Goozee KG, Shah TM, Sohrabi HR, Rainey-Smith SR, Brown B, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016; 115: 449-465. Ref.: https://goo.gl/ebH1tT
- Prakash A, Kumar A. Implicating the role of lycopene in restoration of mitochondrial enzymes and BDNF levels in betaamyloid induced Alzheimers disease. Eur J Pharmacol. 2014; 15: 104-111. Ref.: https://goo.gl/fhMoFc
- Kishi Y, Schmelzer JD, Yao JK, Zollman PJ, Nickander KK, et al. Alpha-lipoic acid: effect on glucose uptake, sorbitol pathway, and energy metabolism in experimental diabetic neuropathy. Diabetes. 1999; 48: 2045-2051. Ref.: https://goo.gl/2BMD7A
- Mitsui Y, Schmelzer JD, Zollman PJ, Mitsui M, Tritschler HJ, et al. Alpha-lipoic acid provides neuroprotection from ischemia-reperfusion injury of peripheral nerve. J Neurol Sci. 1999; 163: 11-16. Ref.: https://goo.gl/BaUXjv
- Hardas SS, Sultana R, Clark AM, Beckett TL, Szweda LI, et al. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol. 2013; 1: 80-85. Ref.: https://goo.gl/obxMQu
- Grundman M. Vitamin E and Alzheimer disease: the basis for additional clinical trials. Am J Clin Nutr. 2000; 71: 630S-636S. Ref.: https://goo.gl/xRQ8px
- Isaac MG, Quinn R, Tabet N. Vitamin E for Alzheimer’s disease and mild cognitive impairment. Cochrane Database Syst Rev. 2008; 3: CD002854. Ref.: https://goo.gl/9KDm16
- Mancuso C, Bates TE, Butterfield DA, Calafato S, Cornelius C, et al. Natural antioxidants in Alzheimer’s disease. Expert Opin Investig Drugs. 2007; 16: 1921-1931. Ref.: https://goo.gl/iPHGWr
- Wattanapenpaiboon N, Wahlqvist MW. Phytonutrient deficiency: the place of palm fruit. Asia Pac J Clin Nutr. 2003; 12: 363-368. Ref.: https://goo.gl/4uKLRi
- Scali C, Giovannini MG, Prosperi C, Bellucci A, Pepeu G, et al. The selective cyclooxygenase-2 inhibitor rofecoxib suppresses brain inflammation and protects cholinergic neurons from excitotoxic degeneration in vivo. Neuroscience. 2003; 117: 909-919. Ref.: https://goo.gl/8MzFFu
- Christen Y. Ginkgo biloba and neurodegenerative disorders. Front Biosci. 2004; 1: 3091-3104. Ref.: https://goo.gl/HVh2aY
- Wang JZ, Wang ZF. Role of melatonin in Alzheimer-like neurodegeneration. Acta Pharmacol Sin. 2006; 27: 41-49. Ref.: https://goo.gl/uQ2MPK
- De la Monte SM. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs. 2012; 72: 49-66. Ref.: https://goo.gl/4bvq9R
- (a) Freiherr J, Hallschmid M, Frey WH, Brunner YF, Chapman CD, et al. Intranasal insulin as a treatment for Alzheimer’s disease: a review of basic research and clinical evidence. CNS Drugs. 2013; 27: 505-514. Ref.: https://goo.gl/6D6eq4; (b) Wang T, Xie C, Yu P, Fang F, Zhu J, et al. Involvement of Insulin Signaling Disturbances in Bisphenol A-Induced Alzheimer’s Disease-like Neurotoxicity. Sci Rep. 2017; 7: 7497. Ref.: https://goo.gl/ahbMMW
- Alagiakrishnan K, Sankaralingam S, Ghosh M, Mereu L, Senior P. Antidiabetic drugs and their potential role in treating mild cognitive impairment and Alzheimer’s disease. Discov Med. 2013; 16: 277-286. Ref.: https://goo.gl/WwaQ93
- Chen Y, Zhang J, Zhang B, Gong CX. Targeting insulin signaling for the treatment of Alzheimer’s disease. Curr Top Med Chem. 2016; 16: 485-492. Ref.: https://goo.gl/GYoxpS
- Wendy Noble, Emmanuel Planel, Cindy Zehr, Vicki Olm, Jordana Meyerson, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci.2005;102: 6990-6995. Ref.: https://goo.gl/WavHmz
- Gruninger F. Invited review: Drug development for tauopathies. Neuropathol Appl Neurobiol. 2015; 41: 81-96. Ref.: https://goo.gl/EDseLe
- Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology. 2014; 76: 27-50. Ref.: https://goo.gl/LMx4TR
- Arai H, Suzuki H, Yoshiyama T, Lobello K, Peng Y, et al. Safety, tolerability and immunogenicity of an immunotherapeutic vaccine (vanutide cridificar [ACC-001]) and the QS-21 adjuvant in Japanese individuals with mild-to-moderate Alzheimer’s disease: a phase IIa, multicenter, randomized, adjuvant and placebo clinical trial. Alzheimer’s Dement. 2013; 9: 282. Ref.: https://goo.gl/o92bKg
- Gong CX, Liu F, Wu G, Rossie S, Wegiel J, et al. Dephosphorylation of microtubule‐associated protein tau by protein phosphatase 5. J Neurochem. 2004; 88: 298-310. Ref.: https://goo.gl/88hoHq
- Fei Liu, Khalid Iqbal, Inge Grundke-Iqbal, Sandra Rossie, Cheng-Xin Gong. Dephosphorylation of Tau by Protein Phosphatase 5 impairment in Alzheimer's disease. J Biological Chemistry. 2005; 280: 1790-1796. Ref.: https://goo.gl/ZMF5yN
- Wilkinson D, Windfeld K, Colding-Jorgensen E. Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2014; 13: 1092-1099. Ref.: https://goo.gl/V2zPw1
- Schneider LS. Idalopirdine for Alzheimer’s disease: written in the stars. Lancet Neurol. 2014; 13: 1063-1065. Ref.: https://goo.gl/6w1bz9
- Marcos B, Chuang TT, Gil-Bea FJ, Ramirez MJ. Effects of 5-HT6 receptor antagonism and cholinesterase inhibition in models of cognitive impairment in the rat. Br J Pharmacol. 2008; 155: 434-440. Ref.: https://goo.gl/r1zzkN
- Wilkinson D, Colding-Jorgensen E, Windfeld K. A clinical phase II study of LU AE58054 added to stable donepezil treatment in patients with moderate Alzheimer’s disease. Alzheimer’s Dement. 2013; 9: 529. Ref.: https://goo.gl/KoukT8
- Maher-Edwards G, Zvartau-Hind M, Hunter AJ, Gold M, Hopton G, et al. Double-blind, controlled phase II study of a 5-HT6 receptorantagonist, SB-742457, in Alzheimer’s disease. Curr Alzheimer Res. 2010; 7: 374-385. Ref.: https://goo.gl/KbDMRV
- Esbenshade TA, Browman KE, Miller TR, Krueger KM, Komater-Roderwald V, et al. Pharmacological properties and procognitive effects of ABT-288, a potent and selective histamine H3 receptor antagonist. J Pharmacol Exp Ther. 2012; 343: 233-245. Ref.: https://goo.gl/5SFdwH
- Othman AA, Haig G, Florian H, Locke C, Gertsik L, et al. The H3 antagonist ABT-288 is tolerated at significantly higher exposures in subjects with schizophrenia than in healthy volunteers. Br J Clin Pharmacol. 2014; 77: 965-974. Ref.: https://goo.gl/Rx2Gsc
- Haig GM, Pritchett Y, Meier A, Othman AA, Hall C, et al. A randomized study of H3 antagonist ABT-288 in mild-to-moderate Alzheimer’s dementia. J Alzheimers Dis. 2014; 42: 959-971. Ref.: https://goo.gl/P4EnRN
- Grove RA, Harrington CM, Mahler A, Beresford I, Maruff P, et al. A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer’s disease. Curr Alzheimer Res. 2014; 11: 47-58. Ref.: https://goo.gl/SqH2B7
- Burstein AH, Grimes I, Galasko DR, Aisen PS, Sabbagh M, et al. Effect of TTP488 in patients with mild to moderate Alzheimer’s disease. BMC Neurol. 2014; 14: Ref.: https://goo.gl/mPxNah
- Prickaerts J, van Goethem NP, Chesworth R, Shapiro G, Boess FG, et al. EVP-6124, a novel and selective alpha7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of alpha7 nicotinic acetylcholine receptors. Neuropharmacology. 2012; 62: 1099-1110. Ref.: https://goo.gl/TqXiGm
- Lawlor B, Kennelly S, O’Dwyer S, Cregg F, Walsh C, et al. NILVAD protocol: a European multicentre double-blind placebo-controlled trial of nilvadipine in mild-to-moderate Alzheimer’s disease. BMJ Open. 2014; 4. Ref.: https://goo.gl/95ZEUd
- Elias Akoury, Michal Gajda, Marcus Pickhardt, Jacek Biernat, Pornsuwan Soraya, et al. Inhibition of Tau Filament Formation by Conformational Modulation. J Am Chem Soc. 2013; 135: 2853-2862. Ref.: https://goo.gl/pYQwZt
- Pul R, Dodel R, Stangel M. Antibody-based therapy in Alzheimer's disease. Expert Opin Biol Ther. 2011; 11: 343-357. Ref.: https://goo.gl/xMvY13
- Bin Zhang, Arpita Maiti, Sharon Shively, Fara Lakhani, Gaye McDonald-Jones, et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci. 2005; 102: 227-231. Ref.: https://goo.gl/5tCRDW
- Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, et al. Microtubule-binding drugs offset tau equestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci. 2005; 102: 227-231. Ref.: https://goo.gl/rF4cFo
- Jama JW, Launer LJ, Witteman JC, den Breeijen JH, Breteler MM, et al. Dietary antioxidants and cognitive function in a population-based sample of older persons. The Rotterdam Study. Am J Epidemiol. 1996; 144: 275-280. Ref.: https://goo.gl/jS9FQi
- Sonnen JA, Larson EB, Gray SL, Wilson A, Kohama SG, et al. Free radical damage to cerebral cortex in Alzheimer's disease, microvascular brain injury, and smoking. Ann Neurol. 2009; 65: 226-229. Ref.: https://goo.gl/DDHV1A
- Ahlemeyer B, Huhne R, Krieglstein J. Retinoic acid potentiated the protective effect of NGF against staurosporine-induced apoptosis in cultured chick neurons by increasing the trkA protein expression. J Neurosci Res. 2000; 60: 767-778. Ref.: https://goo.gl/FSNvaE
- Karasinska JM, Rinninger F, Lutjohann D, Ruddle P, Franciosi S, et al. Specific loss of brain ABCA1 increases brain cholesterol uptake and influences neuronal structure and function. J Neurosci. 2009; 29: 3579-3589. Ref.: https://goo.gl/9XDtn2
- Koldamova RP, Lefterov IM, Ikonomovic MD, Skoko J, Lefterov PI, et al. 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid-β secretion. J Biol Chem. 2003; 278: 13244-13256. Ref.: https://goo.gl/1FK1WR
- Satoh J, Kuroda Y. Amyloid precursor protein β-secretase (BACE) mRNA expression in human neural cell lines following induction of neuronal differentiation and exposure to cytokines and growth factors. Neuropathology. 2000; 20: 289-296. Ref.: https://goo.gl/ohftRG
- Yang Y, Quitschke WW, Brewer GJ. Up regulation of amyloid precursor protein gene promoter in rat primary hippocampal neurons by phorbol ester, IL-1 and retinoic acid, but not by reactive oxygen species. Molecular Brain Research. 1998; 60: 40-49. Ref.: https://goo.gl/1LQEEa
- Wang C, Ward ME, Chen R, Liu K, Tracy TE, et al. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Reports. 2017; 9: 1221-1233. Ref.: https://goo.gl/heqDPE
- Guest J, Garg M, Bilgin A, Grant R. Relationship between central and peripheral fatty acids in humans. Lipids Health Dis. 2013; 12: 79. Ref.: https://goo.gl/gzJdnD
- Barberger-Gateau P, Samieri C, Féart C, Plourde M. Dietary omega 3 polyunsaturated fatty acids and Alzheimer’s disease: interaction with apolipoprotein E genotype. Curr Alzheimer Res. 2011; 8: 479-491. Ref.: https://goo.gl/3YmKjP
- Pan Y, Khalil H, Nicolazzo JA. The Impact of Docosahexaenoic Acid on Alzheimer's Disease: Is There a Role of the Blood-Brain Barrier?. Curr Clin Pharmacol. 2015; 10: 222-241. Ref.: https://goo.gl/4Y7U19
- Chakrabarti M, McDonald AJ, Will Reed J, Moss MA, Das BC, et al. Molecular Signaling Mechanisms of Natural and Synthetic Retinoids for Inhibition of Pathogenesis in Alzheimer's Disease. J Alzheimer’s Dis. 2016; 50: 335-352. Ref.: https://goo.gl/CeE2bd